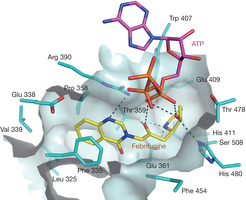
Febrifugine is the active component of the Chinese herb Chang Shan (Dichroa febrifuga Lour.)1, 2, which has been used for treating malaria-induced fever for about 2,000 years. Halofuginone (HF), the halogenated derivative of febrifugine, has been tested in clinical trials for potential therapeutic applications in cancer and fibrotic disease3, 4, 5, 6. Recently, HF was reported to inhibit TH17 cell differentiation by activating the amino acid response pathway7, through inhibiting human prolyl-transfer RNA synthetase (ProRS) to cause intracellular accumulation of uncharged tRNA8, 9. Curiously, inhibition requires the presence of unhydrolysed ATP. Here we report an unusual 2.0 Å structure showing that ATP directly locks onto and orients two parts of HF onto human ProRS, so that one part of HF mimics bound proline and the other mimics the 3′ end of bound tRNA. Thus, HF is a new type of ATP-dependent inhibitor that simultaneously occupies two different substrate binding sites on ProRS. Moreover, our structure indicates a possible similar mechanism of action for febrifugine in malaria treatment. Finally, the elucidation here of a two-site modular targeting activity of HF raises the possibility that substrate-directed capture of similar inhibitors might be a general mechanism that could be applied to other synthetases.





