Like many humans, non-human primates deposit copious misfolded Aβ protein in the brain as they age. Nevertheless, the complete behavioral and pathologic phenotype of Alzheimer’s disease, including Aβ plaques, neurofibrillary (tau) tangles, and dementia, has not yet been identified in a non-human species. Recent research suggests that the crucial link between Aβ aggregation and tauopathy is somehow disengaged in aged monkeys. Understanding why Alzheimer’s disease fails to develop in species that are biologically proximal to humans could disclose new therapeutic targets in the chain of events leading to neurodegeneration and dementia.
Trends
The accumulation of misfolded, multimeric Aβ and tau proteins in the brain is a defining pathologic feature of AD, the leading cause of dementia in humans.
Senescent non-human primates deposit abundant Aβ in senile plaques and cerebral amyloid angiopathy, and mild intracellular hyperphosphorylated tau is sometimes present; however, the AD triad of significant senile plaques, neurofibrillary tangles, and dementia has not yet been identified in a non-human species.
Establishing why species that are biologically similar to humans are resistant to AD could reveal novel mechanistic objectives for the potential development of preventive measures and/or treatments.
Alzheimer’s Disease: A Uniquely Human Disorder?
The more similar animals are biologically, the more likely they are to manifest similar diseases. The full phenotype of Alzheimer’s disease (AD; see Glossary), however, has not yet been found in any non-human species [1, 2, 3, 4, 5]. Recent research has reinforced the view that non-human primates – our closest biological relatives – are surprisingly resistant to an AD-like disorder as they grow old, despite the occurrence of AD-linked genetic, molecular, and cellular mechanisms that are similar to those in humans [5, 6, 7, 8].
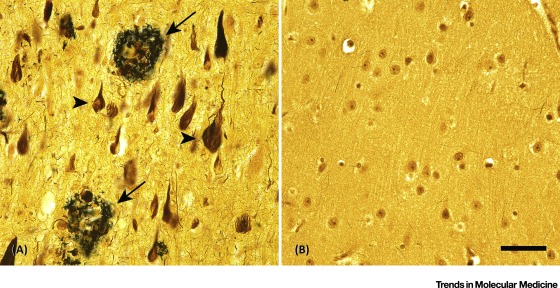
Central to the pathogenesis of AD is the aggregation, proliferation, and spread of two misfolded proteins in the brain: Aβ and the microtubule-associated protein tau (MAPT) [9]. The aberrant proteins propagate by a prionlike process [10] that is evident histologically in the presence of extracellular Aβ plaques, cerebral Aβ-amyloid angiopathy (CAA) [11], and intracellular neurofibrillary tangles and neuropil threads consisting of hyperphosphorylated tau [12, 13, 14]. The relationship of histopathologic changes per se to dementia remains somewhat ambiguous, 影梭 but a definitive diagnosis of AD requires the significant presence of plaques and tangles (Figure 1) in a patient with clinical signs and symptoms of cognitive change that typify dementia [15]. Indeed, both tauopathy and Aβ proteopathy are necessary for the complete clinicopathologic expression of AD [16], but tauopathy is a particularly potent driver of neuronal dysfunction and cognitive decline [17, 18, 19, 20, 21]. Genetic and biomarker evidence, however, pinpoints the accumulation of misfolded Aβ as the earliest obligatory event in the ontogeny of AD [9, 22].
Paradoxically, aged monkeys and apes can accumulate large quantities of Aβ in the brain, yet in the species that have been analyzed, AD-like tangles are rare or absent (Figure 2). Furthermore, despite some decline in behavioral capabilities, a profound, dementia-like disorder has not been discovered in a non-human primate [2]. Aged dogs can also develop a degree of cognitive dysfunction and Aβ deposition, but they do not present neurofibrillary tangles in the brain [23, 24]. In this review article, we describe the comparative neurobiology of aging in humans and non-human primates, and then discuss how understanding this critical discrepancy – Aβ plaques in the absence of tangles – could yield insights into the cascade of events leading from Aβ aggregation to tauopathy and dementia in humans (Box 1)……





