Another brick in the wall against herpes
A wide variety of host immune defenses are mobilized to contain human infection with the HSV-1 herpesvirus. Herpes simplex encephalitis (HSE) is a devastating form of HSV-1 infection that can indicate an underlying host immunodeficiency. Using whole-exome sequencing to search for a genetic etiology of HSE in a young boy, Naesens et al. discovered hypofunctional mutations in the GTF3A gene encoding the TFIIIA subunit of RNA polymerase III as the cause of impaired innate immunity to HSV-1. TFIIIA-mediated transcription of a host-derived noncoding ribosomal RNA contributes to activation of the RIG-I cytoplasmic RNA sensor and a type I interferon response contributing to HSV-1 control. These findings reveal another critical layer of innate defense that human cells use to control HSV-1 herpesvirus infections.
Abstract
Herpes simplex virus 1 (HSV-1) infects several billion people worldwide and can cause life-threatening herpes simplex encephalitis (HSE) in some patients. Monogenic defects in components of the type I interferon system have been identified in patients with HSE, emphasizing the role of inborn errors of immunity underlying HSE pathogenesis. Here, we identify compound heterozygous loss-of-function mutations in the gene GTF3A encoding for transcription factor IIIA (TFIIIA), a component of the RNA polymerase III complex, in a patient with common variable immunodeficiency and HSE. Patient fibroblasts and GTF3A gene–edited cells displayed impaired HSV-1–induced innate immune responses and enhanced HSV-1 replication. Chromatin immunoprecipitation sequencing analysis identified the 5S ribosomal RNA pseudogene 141 (RNA5SP141), an endogenous ligand of the RNA sensor RIG-I, as a transcriptional target of TFIIIA. GTF3A mutant cells exhibited diminished RNA5SP141 expression and abrogated RIG-I activation upon HSV-1 infection. Our work unveils a crucial role for TFIIIA in transcriptional regulation of a cellular RIG-I agonist and shows that GTF3A genetic defects lead to impaired cell-intrinsic anti–HSV-1 responses and can predispose to HSE.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) is one of the most common human pathogens, infecting several billion people worldwide (1). HSV-1 is primarily transmitted through oral-to-oral contact and commonly causes mild mucocutaneous infections. However, in rare cases (1 in 250,000 to 500,000 individuals per year), the virus causes herpes simplex encephalitis (HSE) predominantly in children younger than 3 years old (2). Late diagnosis of HSE or lack of adequate antiviral therapy is associated with high mortality (~70%), and most survivors develop neurological sequelae (2).
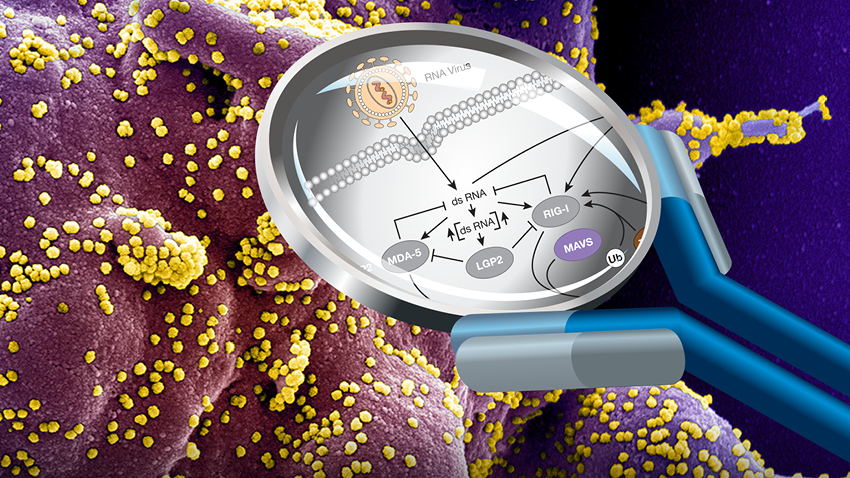
The host innate immune surveillance machinery plays a crucial role in the defense against HSV-1 and dictates the outcome of infection (3). Nucleic acid sensors survey different cellular compartments for viral or cellular RNA or DNA ligands and induce the production of type I interferons (IFN-I), proinflammatory cytokines, and chemokines (4). Studies in cell culture and/or in mice have shown that different classes of nucleic acid receptors contribute to the sensing of HSV-1 infection, including Toll-like receptors (e.g., TLR3 and TLR9) and the intracellular sensors cGAS and IFI16 (5). Human studies demonstrated the importance of the double-stranded RNA (dsRNA) sensor TLR3 in restricting HSV-1 replication and pathogenesis by mediating constitutive IFN-I immunity (6, 7). Accumulating evidence from in vitro experiments using a variety of cell types showed that the cytoplasmic RNA sensor RIG-I, which signals through MAVS (mitochondrial antiviral signaling protein), also critically contributes to the innate immune response to HSV-1 and other herpesviruses such as varicella-zoster virus (VZV), Epstein-Barr virus (EBV), and Kaposi’s sarcoma–associated herpesvirus (KSHV) (8, 9). RNA polymerase III (Pol III) has been shown to convert host- or virus-derived DNA products into 5′-triphosphate RNA ligands for RIG-I (10, 11). RIG-I affinity purification from HSV-1–infected cells and next-generation sequencing of bound RNA revealed that small noncoding RNAs, in particular the 5S ribosomal pseudogene transcript RNA5SP141 (5S ribosomal RNA pseudogene 141), serve as RIG-I cellular ligands (12).
Inborn errors in genes implicated in TLR3 nucleic acid sensing or in downstream signaling components (UNC93B1, TLR3, TRIF, TRAF3, TBK1, or IRF3) account for ~5% of HSE cases (7). Two recent studies revealed genetic defects in TLR3-independent anti-herpesviral mechanisms, including autosomal recessive DBR1 deficiency and heterozygous mutations in SNORA31 for which the molecular antiviral mechanism(s) remains to be elucidated (13, 14). To date, for many HSE cases, the underlying genetic defects are still unknown.
Here, we identify a role for loss-of-function (LOF) mutations in GTF3A, encoding for transcription factor IIIA (TFIIIA), which is part of the Pol III complex, in HSE. Our work shows that TFIIIA induces the expression of the RIG-I ligand RNA5SP141, thereby enabling protective IFN-I–mediated immunity against HSV-1 infection, which is abrogated in cells harboring GTF3A mutations….





