One means by which cancer cells evade therapies involves their ability to reprogram to a cell type that no longer depends on the cellular pathway being targeted by the treatments. Hormone deprivation therapies that suppress androgen receptor (AR) signaling are the mainstay of treatment for metastatic prostate cancer. However, prostate cancers can become resistant to this approach by losing dependence on androgen hormones. On pages 84 and 78 of this issue, Mu et al. (1) and Ku et al. (2), respectively, contribute to our mechanistic understanding of this remarkable plasticity in cell identity, which allows cancers to thrive.
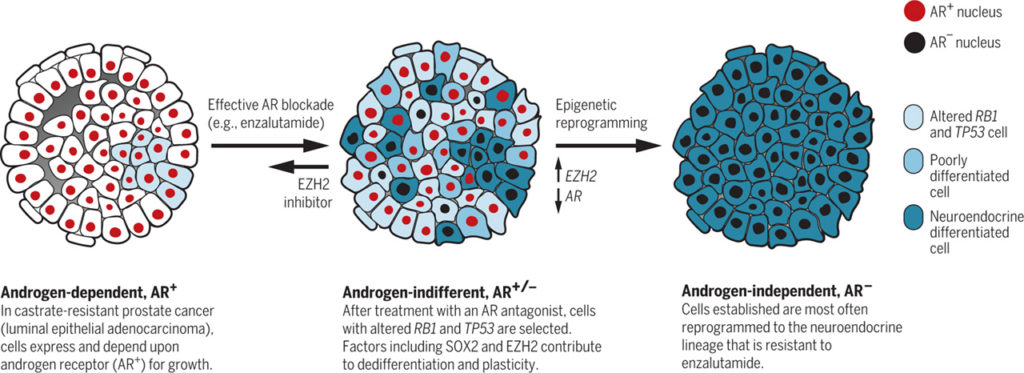
Androgens stimulate prostate cancer cell growth. The main androgens are testosterone and dihydrotestosterone, which are synthesized primarily in the testes. Decreasing androgen production or preventing the hormones from acting on prostate cancer cells often makes the tumors shrink or grow more slowly. However, prostate cancer can adapt to androgen deprivation through alterations that restore AR signaling and maintain their luminal epithelial adenocarcinoma phenotype, even when androgen production is low [referred to as castration-resistant prostate cancer-adeno (CRPC-adeno)] (3). With the development of more effective AR-targeting drugs such as abiraterone and enzalutamide, additional resistance mechanisms are arising. About a quarter of these resistant tumors undergo cellular reprogramming and acquire a continuum of neuroendocrine characteristics (CRPC-NE) (4, 5). Genomic analyses have shown that CRPC-NE evolves from CRPC-adeno. Most CRPC-NE express one or more NE-lineage markers [such as synaptophysin (SYP)], and there are a range of morphological variants, perhaps reflecting variable differentiation states. The increased expression of AR and AR-regulated genes is generally reduced in CRPC-NE compared to CRPC-adeno, although there is a range of overlap that may reflect ongoing selection as well as differences in genomics (6).
In addition to NE-lineage markers, the messenger RNA profiles (transcriptomes) of CRPC-NE patient samples and prostate cancer models have shown increased expression of genes involved in neuronal development, such as sex determining region Y box 2 (SOX2), and genes encoding epigenetic regulators, such as enhancer of zeste homology 2 (EZH2) and DNA methyl-transferase 1 (DNMT1) (6, 7). DNMT1 may contribute to epigenetic characteristics, such as DNA methylation patterns, that are markedly different between CRPC-adeno and CRPC-NE (6). In the most comprehensive genomic analysis of CRPC-NE to date (6), the co-occurrence of alterations in cell signaling pathways involving the tumor suppressor proteins retinoblastoma 1 (RB1) and tumor protein 53 (TP53) was highly enriched in CRPC-NE (∼50%) relative to CRPC-adeno (ů15%), suggesting the involvement of these pathways in the selection of CRPC-NE. Mu et al. and Ku et al. connect the loss of the RB1 and TP53 genes to lineage plasticity and epigenetic regulation in prostate cancer resistance to androgen deprivation therapy.
Mu et al. addressed the phenotypic consequences of RB1 and TP53 silencing in a human cell line that overexpresses AR (LNCaP-AR cells), a model of CRPC-adeno that is sensitive to the AR antagonist enzalutamide. Silencing both RB1 and TP53, but neither alone, caused marked enzalutamide resistance in these cells, although AR activity persisted and remained responsive to enzalutamide. Notably, cells lacking TP53 and RB1 displayed lineage plasticity, as indicated by decreased expression of luminal epithelial cell markers and increased expression of basal epithelial cell and neuroendocrine markers. Moreover, these gene expression changes occurred within 48 hours of induced depletion of TP53 and RB1 and could be rapidly reversed, indicating a direct effect rather than selection for cells with preexisting phenotypic differences. Mu et al. used RNA-sequencing data sets of clinical samples to identify transcription factors whose expression correlated with co-occurring alterations in the RB1 and TP53 signaling pathways. Among these, SOX2 expression was induced by silencing RB1 and TP53 in LNCaP-AR cells, and SOX2 expression in these cells was necessary and sufficient for the expression of basal epithelial and neuroendocrine markers as well as for enzalutamide resistance. RB1 loss has been associated with lineage plasticity in other cancer cell models, and in induced pluripotent stem cells, RB1 inhibited the basal transcription of SOX2 and other pluripotency genes (8). These data suggest a model whereby the induction of SOX2 expression subsequent to RB1 and TP53 loss contributes to neuroendocrine differentiation and AR-pathway independence…….





