Abstract
Sulfonylureas are anti-diabetic medications that act by inhibiting pancreatic KATP channels composed of SUR1 and Kir6.2. The mechanism by which these drugs interact with and inhibit the channel has been extensively investigated, yet it remains unclear where the drug binding pocket resides. Here, we present a cryo-EM structure of a hamster SUR1/rat Kir6.2 channel bound to a high-affinity sulfonylurea drug glibenclamide and ATP at 3.63 Å resolution, which reveals unprecedented details of the ATP and glibenclamide binding sites. Importantly, the structure shows for the first time that glibenclamide is lodged in the transmembrane bundle of the SUR1-ABC core connected to the first nucleotide binding domain near the inner leaflet of the lipid bilayer. Mutation of residues predicted to interact with glibenclamide in our model led to reduced sensitivity to glibenclamide. Our structure provides novel mechanistic insights of how sulfonylureas and ATP interact with the KATP channel complex to inhibit channel activity.
https://doi.org/10.7554/eLife.31054.001
Introduction
ATP-sensitive potassium (KATP) channels are unique hetero-octameric complexes each composed of four inwardly rectifying Kir6 channel subunits and four sulfonylurea receptor (SUR) subunits belonging to the ATP binding cassette (ABC) transporter protein family (Aguilar-Bryan and Bryan, 1999; Nichols, 2006). In pancreatic β-cells, KATP channels formed by Kir6.2 and SUR1 are gated by intracellular ATP and ADP, with ATP inhibiting channel activity while Mg2+-complexed ATP and ADP stimulating channel activity (Aguilar-Bryan and Bryan, 1999; Ashcroft, 2007). During glucose stimulation, the intracellular ATP to ADP ratio increases following glucose metabolism, which favors channel closure by ATP, resulting in membrane depolarization, Ca2+ influx, and exocytosis of insulin granules. In this way, KATP channels are able to control insulin secretion according to blood glucose levels. Mutations that disrupt channel function are known to cause a spectrum of insulin secretion disorders (Ashcroft, 2005; Koster et al., 2005a). Specifically, loss-of-function mutations result in congenital hyperinsulinism, whereas gain-of-function mutations lead to transient or permanent neonatal diabetes (Ashcroft, 2005). The pivotal role of KATP channels in insulin secretion regulation makes them an important drug target.
Discovered in the 1940s, sulfonylureas have been a mainstay of type 2 diabetes therapy for more than half a century (Sola et al., 2015). The medical importance of this class of drugs has led to its evolution into several generations of agents, including first-generation sulfonylureas such as tolbutamide and second-generation agents such as the high-affinity sulfonylurea glibenclamide (GBC) (Gribble and Reimann, 2003; Sola et al., 2015). All sulfonylureas stimulate insulin secretion to reduce plasma glucose levels by inhibiting the activity of β-cell KATP channels (Gribble and Reimann, 2003). More recently, they have also become the primary pharmacotherapy for neonatal diabetes patients carrying gain-of-function KATP channel mutations (Aguilar-Bryan and Bryan, 2008; Ashcroft, 2007; Sagen et al., 2004). Despite their clinical importance and decades of research, how sulfonylureas interact with and inhibit KATP channel activity remains poorly understood.
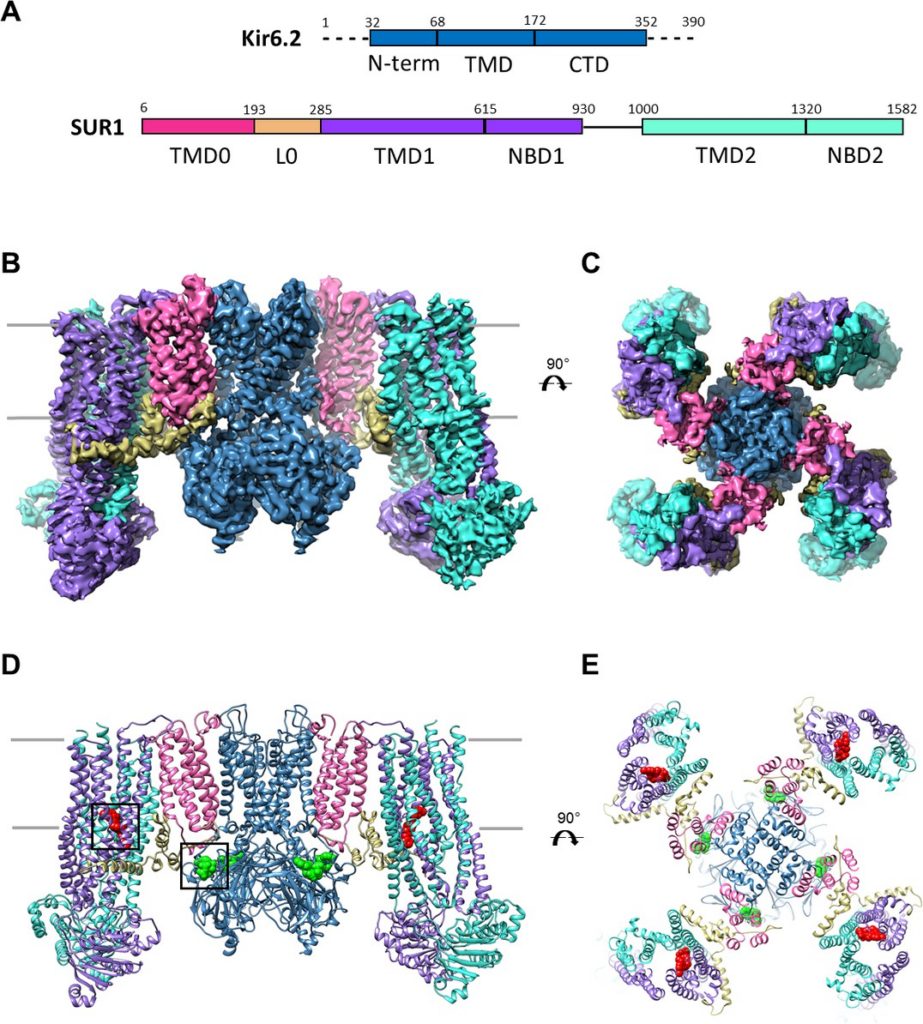
To begin to address the structural mechanisms by which ATP and sulfonylureas such as GBC inhibit KATP channels to stimulate insulin secretion, we recently carried out single particle cryo-EM and determined the structure of the β-cell KATP channel complex in the presence of ATP and GBC (Martin et al., 2017). While our initial structure at a resolution of 5.7 Å revealed the overall architecture of the channel and location of the ATP molecule, it was unable to clearly define the GBC binding site and the atomic details associated with ATP binding. A concurrent study by Li et al. (Li et al., 2017) reported another cryo-EM KATP channel structure at 5.6 Å resolution, also in the presence of GBC but without ATP, in which the GBC binding site was proposed to lie near the cytoplasmic linker between the first and second transmembrane domains of SUR1; however, the assignment of the GBC density was tentative. To resolve the binding sites for ATP and GBC, we performed additional studies and improved the resolution of the KATP channel structure bound to GBC and ATP to ~3.6 Å. The higher resolution structure not only clearly defines the GBC and ATP binding pockets but also provides novel insights into the mechanisms of channel inhibition by ATP and GBC.





