Most viral infections trigger B cells to produce neutralizing antibodies, but for a handful of viruses that cause chronic infections, B cells are for some reason unable to do their job. The biggest driver for this suppression, at least for the mouse lymphocytic choriomeningitis virus (LCMV), turns out to be a group of cytokines collectively called type I interferon (IFN-I), according to three independent reports published today (October 21) in Science Immunology.
“All of the three papers identified type I IFN as the culprit,” said Matteo Iannacone, an immunologist at the San Raffaele Scientific Institute in Milan who coauthored one of the studies.
For decades, immunologists have used LCMV as a model system to study T cell–dominant immune responses because B cell production of virus-neutralizing antibodies is absent or weak and delayed for several weeks. But because some human viruses, such as HIV and hepatitis B (HBV), also fail to induce strong antibody responses, Dorian McGavern, a viral immunologist at the National Institute of Neurological Disorders and Stroke, and others decided to use LCMV to find mechanisms behind B-cell failure.
McGavern’s team started by injecting uninfected mice with B cells engineered to recognize LCMV, then exposed them to the virus. Within a week, the injected B cells disappeared from the animals’ spleens. If the researchers waited until six days after infection to inject the B cells, however, the cells stuck around. In a separate study, Daniel Pinschewer, a virologist at the University of Basel similarly noticed a three-day “death zone” for LCMV-specific B cells.
Both teams knew LCMV induces systemic production of high levels of IFN-I during the first few days of infection. “Type I IFN is the most important initiator of the alarm bell,” said McGavern. “It is what drives immunity to viruses.” But in the case of LCMV, the cytokine seems to have a bad side. When both teams blocked the IFN-I receptor before infection, numbers of LCMV-specific B cells shot up in the spleens of infected mice, allowing the animals to produce significantly higher levels of virus-neutralizing antibodies.
To determine what caused the loss of B cells in response to IFN-I signaling, Pinschewer’s team infected several types of knockout mice and found that myeloid cells, dendritic cells, and T cells were all involved.
McGavern and his colleagues found that CD8+ T cells responded to IFN-I by killing off LCMV-specific B cells within the first few days of infection. Because CD8+ T cells typically take at least a week to respond to infection, McGavern said it was surprising to see them acting so early, but his team confirmed it by recording interactions between B and T cells in spleens and lymph nodes of living mice. “You have direct visual evidence that cytotoxic T lymphocytes can touch and kill B cells,” he said.
According to McGavern’s study, the key to losing LCMV-specific B cells is infection through the B cell receptor, which binds and internalizes the virus. CD8+ T cells then kill the infected B cells, eliminating their chance to make antibodies. “If a virus has the potential to gain access to a B cell and infect it upon being pulled in, now that subjects that cell to killing,” he said.
But, Steven Waggoner, an immunologist at the University of Cincinnati College of Medicine who was not involved in any of the studies, told The Scientist that LCMV does not typically infect B cells through the B cell receptor, so it’s still unclear why CD8+ T cells would preferentially target B cells that have LCMV-specific receptors.
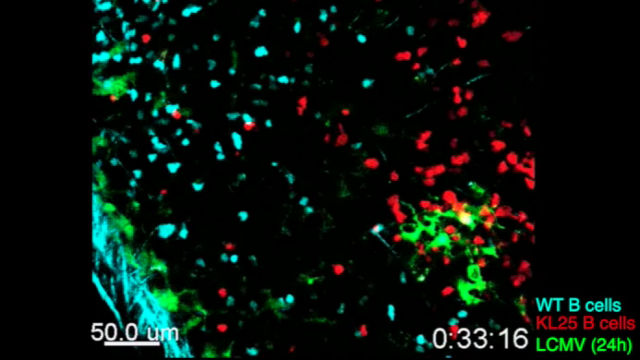
In a third study, Iannacone and his team injected mice with fluorescently labeled B cells that were specific to LCMV or to vesicular stomatitis virus (VSV), which is known to induce strong antibody responses. They then infected the mice through the skin with the corresponding virus, and recorded the B cells’ movements within a nearby lymph node.
The team found that after infection, both B cell types leave the lymph node follicles, where B cells divide, to interact with infected cells in other areas of the lymph node. But only VSV-specific B cells returned to the follicles, where they went on to mature and make neutralizing antibodies. LCMV-specific B cells remained outside the follicle for at least three days, and instead interacted with nearby monocytes.
The researchers found that the monocytes killed the B cells by secreting a protein called nitric oxide synthase. Once again, IFN-I appeared to play a key role in directing this cellular killing. In mice deficient for the IFN-I receptor, monocytes could not migrate to the lymph nodes after LCMV infection and B cells were spared.
In contrast to McGavern’s work, Iannacone’s team was unable to find a role for CD8+ T cells in B cell deletion. Likewise, McGavern found no role for monocytes, and said the routes and timing of the two infection models likely accounts for the difference. Iannacone said IFN-I may affect immune cells differently at different stages or locations of infection.
“There’s so much downstream of IFN-I,” Pinschewer said. “It’s probably the combined action of a plethora of factors.”
According to Pinschewer, this new suite of evidence for IFN-I’s role in suppressing LCMV B cell responses should be considered in the contexts of other chronic infections, such as HIV, HBV, and tuberculosis. Waggoner agreed: “You could take this to a couple of preclinical models and show relevance to human pathogens.”
However, Pinschewer said, because IFN-I is so vital for virus control, researchers will have to find ways to eliminate its negative affects without losing its benefits. “I don’t think we would like to deplete type I IFN as a therapeutic.”





