Abstract
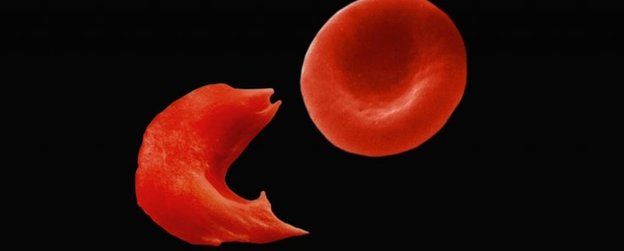
Sickle cell disease is a growing health burden afflicting millions around the world. Clinical observation and laboratory studies have shown that the severity of sickle cell disease is ameliorated in individuals who have elevated levels of fetal hemoglobin. Additional pharmacologic agents to induce sufficient fetal hemoglobin to diminish clinical severity is an unmet medical need. We recently found that up-regulation of peroxisome proliferator–activated receptor γ coactivator-1α (PGC-1α) can induce fetal hemoglobin synthesis in human primary erythroblasts. Here, we report that a small molecule, SR-18292, increases PGC-1α leading to enhanced fetal hemoglobin expression in human erythroid cells, β-globin yeast artificial chromosome mice, and sickle cell disease mice. In SR-18292–treated sickle mice, sickled red blood cells are significantly reduced, and disease complications are alleviated. SR-18292, or agents in its class, could be a promising additional therapeutic for sickle cell disease.
INTRODUCTION
Sickle cell disease (SCD) is caused by a single missense mutation in the adult β-globin gene that codes for sickle hemoglobin (HbS; α2βS2). Deoxygenated HbS tends to form polymers that damage the red blood cell (RBC) causing hemolytic anemia, vaso-occlusion, and acute and chronic pain with progressive multiorgan damage, all of which lead to early mortality (1–3). Clinical, epidemiologic, and genetic studies have shown that sufficient reactivation of fetal hemoglobin (HbF; α2γ2) alleviates the symptoms of SCD because HbF dilutes HbS and directly disrupts its polymerization (4–6). Identifying effective means of increasing HbF in erythroid progenitors has been pursued for many decades (7). Although the Food and Drug Administration (FDA) has approved three new drugs to treat SCD, none target HbF. Voxelotor inhibits deoxy-HbS polymerization by increasing hemoglobin-oxygen affinity; l-glutamine and crizanlizumab target SCD pathophysiology downstream of deoxy-HbS polymerization (8). Currently, the standard of care for treating SCD is hydroxyurea (HU), which is the only FDA-approved HbF-inducing drug. However, adult patients with SCD rarely respond to HU with HbF levels approaching those attained in young children that can have a profound effect on their disease (9). Other reported that HbF inducers like 5-azacytidine, 5-aza-2′-deoxycytidine, and butyrate have limited effectiveness or have been toxic (4, 10). Additional efficacious, safe, and widely available HbF-inducing drugs that do not expose patients to the rigors of current iterations of cellular therapeutics are an unmet need for treating SCD.
The peroxisome proliferator–activated receptor γ (PPARγ) coactivator-1α, or PGC-1α, was first found in brown fat as a PPARγ-interacting protein (11). PGC-1α belongs to a family of coactivator proteins that are key factors in regulating various signaling pathways (12–15). In addition to its effects on the liver, neurons, and muscle (16–18), recent evidence suggests that PGC-1α also plays an important role in the maturation and survival of erythroid cells. In mice, which do not have fetal globin genes, PGC-1α loss-of-function mutants were stunted and anemic. Expression of their embryonic εy- and βh1-globin genes was reduced. Moreover, PGC-1α initiated globin gene transcriptional activation by interacting with nuclear receptors testicular receptor 2/4 (TR2/TR4) (19, 20). Forced overexpression of PGC-1α in vitro by adenovirus infection in bone marrow cells from SCD mice resulted in significantly increased expression of human fetal γ-globin genes (HBG1 and HBG2, together referred to as HBG) and εy-globin as well as βh1-globin gene, the murine fetal globin analog (21). In human CD34+ cells, ZLN005, a small-molecule PGC-1α activator, can up-regulate PGC-1α and increase HbF synthesis (22).
Here, we show that another small molecular modulator of PGC-1α, SR-18292 (23), increased the expression of PGC-1α and γ-globin genes in human CD34+ hematopoietic stem and progenitor cells. The number of HbF-positive cells (F-cells) derived from CD34+ cells was increased. SR-18292–treated human β-globin yeast artificial chromosome (β-YAC) transgenic mice and SCD mice exhibited increased HBG mRNA expression, HBG protein, and F-cell numbers. Irreversibly sickled RBCs and reticulocytes were significantly reduced in the peripheral blood of SR-18292–treated SCD mice. This was associated with diminished hemolysis, reduced splenomegaly, and less organ necrosis. These results suggest that modulating PGC-1α activity or the signaling pathways that it regulates might therapeutically benefit patients with SCD. Our findings provide initial proof of principle for developing more effective HbF-boosting therapeutics directed at this pathway.
RESULTS
Effects of SR-18292 on human primary erythroid progenitor CD34+ cells
Human CD34+ cells were isolated from the peripheral blood of normal adults and cultured in a three-phase culture system (24). Cells were treated with SR-18292 at phase 1 day 0. After 11 days of SR-18292 treatment, mRNA levels of PGC-1α were significantly increased in treated cells with a 4- to 5-fold increase at the lower doses of 1.0 or 2.5 μM and about 10-fold increase at the higher doses of 5.0 or 7.5 μM SR-18292. Fetal γ-globin (HBG) mRNA levels were increased 2.5- and 4- to 6-fold at the lower and higher doses; adult β-globin (HBB) mRNA levels were unaffected (Fig. 1A). Protein levels of γ-globin determined by Western blot were increased in human CD34+ cells treated with SR-18292 at all tested concentrations (Fig. 1, B and C). Increased γ-globin mRNA levels in treated cells were associated with a significant increase in the percentage of F-cells compared with the untreated or vehicle control–treated cells. SR-18292–treated cells CD34+ cells had F-cell counts between 27.4 ± 0.8% and 48.7 ± 3.0%, while controls had <20% F-cells (Fig. 1, D and E). We next determined whether SR-18292 treatment could affect erythroid differentiation in human CD34+ cells. Erythroid cellular differentiation markers (CD71/CD235a) and terminal differentiation markers (CD49d/CD233) were examined by flow cytometric analyses of cultured CD34+ cells treated with vehicle control or SR-18292 (25). There was no statistically significant difference in the population of CD71+CD235a+ cells among all groups (fig. S1, A and B). However, SR-18292 treatment increased the percentage of CD49d−CD233+ cells at 5.0 and 7.5 μM (75.8 ± 1.1% and 78.6 ± 2.8%, respectively) compared with untreated (49.3 ± 1.4%) or vehicle control groups (44.8 ± 3.2%) (fig. S1, C and D). These results suggest that compared with doses of 1.0 and 2.5 μM, SR-18292 at higher concentrations can promote erythroid terminal differentiation.
To further determine whether PGC-1α up-regulation by SR-18292 can directly account for these HbF-inductive effects, we examined the effect of SR-18292 in human CD34+ cells with PGC-1α knocking down. Human CD34+ cells, treated with or without SR-18292, were infected with adenoviruses carrying PGC-1α short hairpin RNA (shRNA) (pAdEasy-TetO-H1-PGC-1α shRNA) or empty vector control (pAdEasy-TetO-H1). The abundance of PGC-1α mRNA in cells infected with the PGC-1α shRNA virus was significantly decreased as compared to control cells. As a result, the HbF-inductive effect of SR-18292 was only exhibited in noninfected or control virus-infected cells but not in PGC-1α shRNA virus-infected cells (fig. S2). These results demonstrate that the inductive effect of SR-18292 on γ-globin expression and HbF synthesis is dependent on PGC-1α.
Combination of SR-18292 with HU and decitabine
HU is standard of care for treating SCD currently (9). Decitabine (5-aza-2′-deoxycytidine; DAC) can induce HbF expression by inhibiting DNA methyltransferase, which is part of an HBG repressor complex (26). A phase 2 trial evaluating the efficacy and safety of oral decitabine tetrahydrouridine (NDec) (27) in patients with SCD is ongoing (ClinicalTrials.gov, NCT05405114). To examine whether SR-18292 combined with HU or DAC has an additive effect on HbF induction, we studied the effects of combination treatment in human CD34+ cells. Combinations of SR-18292 with HU or DAC had higher levels of γ-globin mRNA (Fig. 2A). SR-18292 at 2.5 μM with 25 μM HU or 0.05 μM DAC was associated with increased F-cells compared with HU alone or DAC alone, with F-cells reaching levels of 60 to 70% (Fig. 2, B and C). The additive effect on HbF induction, when SR-18292 is combined with HU or DAC, is consistent with these compounds having different mechanisms of inducing HbF synthesis, which would therapeutically benefit patients with SCD with less-than-optimal responses to HU or DAC.





