In this issue of JEM, Marciniak et al. (https://doi.org/10.1084/jem.20161731) identify a putative novel function of tau protein as a regulator of insulin signaling in the cartier jewelry replica brain. They find that tau deletion impairs hippocampal response to insulin through IRS-1 and PTEN dysregulation and suggest that, in Alzheimer’s disease, impairment of brain insulin signaling might occur via tau loss of function.
Alzheimer’s disease (AD) is the leading form of dementia worldwide. The two major histopathological hallmarks of AD are senile fake cartier bracelets
plaques composed of amyloid-β (Aβ) peptide and neurofibrillary replica cartier love bracelets tangles made of abnormally hyperphosphorylated tau protein. Tau pathology is important because it correlates with the degree of cognitive impairment in AD patients. The majority of AD cases are late onset and sporadic, and many environmental, biological, and genetic factors are thought to contribute to the disease. Epidemiological studies particularly suggest that metabolic disorders such as type 2 diabetes (T2D) could be such factors, as they are associated with a higher risk of AD later in life.
Brain insulin resistance appears to be an early and common feature of AD (for review see Stanley et al., 2016), and AD has been proposed as a “type 3 diabetes” representing a form of diabetes that selectively involves the brain (de la Monte and Wands, 2008). Our current knowledge on how AD pathologies may alter brain insulin signaling relies on evidence showing the development of brain insulin resistance after oligomeric Aβ exposure, thus implicating amyloid pathology as a major mediator of brain insulin resistance in AD (Bomfim et al., 2012). However, although the impact of insulin dysfunction on tau pathogenesis has been extensively studied (for review see El Khoury et al., 2014), the effects of tau pathology on insulin signaling has never been assessed before.
Tau is a microtubule binding protein whose most well-known function is to bind and stabilize microtubules. But it has also been suggested to have many other functions such as regulation of cell signaling, synaptic plasticity, and genomic stability (Guo et al., 2017). Tau pathology in AD is thought to exert its detrimental effects through a toxic gain of function, but a potential loss of physiological function might also contribute to some phenotype of the disease. In this issue, Marciniak et al. hypothesized that in AD, tau loss of function could alter brain insulin signaling and partly explain the cognitive and metabolic impairments observed in AD.
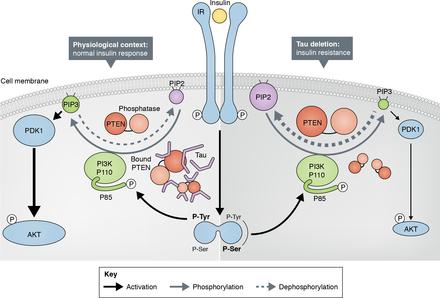
Using mice depleted for MAPT, the tau gene (tau KO mice), Marciniak et al. (2017) initially identified a reduction of hippocampal long-term depression of extracellular field excitatory postsynaptic potentials in brain slices from tau KO mice compared with littermate controls after insulin treatments. Altered response to insulin was confirmed in the same mice model ex vivo and in vivo with decreased activation of IRS-1 and AKT (both implicated in insulin signaling), suggesting brain insulin resistance in tau KO mice. By taking advantage of coimmunoprecipitation experiments and bimolecular fluorescence complementation assay, the authors further evaluated whether tau directly interacts with key insulin signaling molecules in neuroblastoma cells expressing non-mutated human hermes birkin bag tau protein. Unexpectedly, tau did not seem to interact with either the insulin receptor IRS-1 or with PI3K (p85). However, tau was found to interact with PTEN, a phosphatase known to inhibit insulin signaling through the PI3K-Akt pathway. Moreover, Marciniak et al. (2017) demonstrated that human tau is able to reduce PTEN activity and thus promote PIP3 production alone or by potentiating the effect of insulin (see figure)
Next, Marciniak et al. (2017) assessed whether tau deletion alters brain insulin functions. Interestingly, absence of tau reduced the anorexigenic effect of intracerebroventricular injection of insulin in tau KO mice. These mice also developed peripheral hyperinsulinemia and glucose intolerance. These data confirmed an important role of tau in the regulation of energy metabolism. Finally, the authors noticed an effect of tau haplotype on glucose tolerance hermes outlet in published genome-wide association study (GWAS) data. H1 haplotype is associated with higher risk of tauopathies (Pittman et al., 2005), and in the study by Marciniak et al. (2017), patients with H1 haplotype exhibited higher circulating glucose levels and lower insulin levels during an oral glucose tolerance test, suggesting that tau impacts peripheral metabolism in humans. Overall, the in vivo and in vitro results dovetail nicely together and with the GWAS data to provide compelling evidence that tau can regulate both brain insulin signaling and peripheral glucose metabolism.
The study by Marciniak et al. (2017) further addresses a question that has emerged over the last few years as to whether AD is a cause or consequence of insulin signaling impairment (Stanley et al., 2016). Epidemiological studies supported by in vivo and in vitro experiments establish metabolic problems as risks for AD. However, the study by Marciniak et al. (2017) is the first assessing whether tau pathology affects brain insulin signaling in AD. Interestingly, metabolic changes and central insulin resistance have been reported in other tauopathies such as progressive supranuclear palsy or corticobasal degeneration (Ahmed et al., 2014; Yarchoan et al., 2014). This suggests that the alteration of insulin signaling resulting from the loss of tau function upon tau pathology may explain metabolic changes in many tauopathies. At the same time, Marciniak et al. (2017) raise new questions regarding mechanisms underlying the role of tau protein as a regulator of brain insulin signaling that need clarification. For instance, the two molecular events suggested to explain this novel function of tau involved IRS-1 and/or PTEN. Early studies have in fact reported that total IRS-1 but also IRS-2 are decreased in the brain of AD patients along with increased phosphorylated IRS-1 on Ser636/639 and Ser616, which colocalizes and correlates with neurofibrillary tangle deposition and inversely correlates with cognitive score (Ma et al., 2009; Moloney et al., 2010; Talbot et al., 2012). This is consistent with altered IRS-1 activity in tau KO mice reported in this issue, whereas the authors could not establish a direct interaction between tau and IRS-1. Another replica cartier love bracelet explanation lies in the direct interaction between PTEN and tau, which can modulate the PTEN activity and thus explain decreased responsiveness to insulin in tau KO mice. Although at the current stage it is still impossible to determine which of IRS-1 or PTEN is the instrument of tau to regulate brain insulin signaling, the intervention of these two proteins together in this process is not to be neglected, especially considering that PTEN has been shown to act as tyrosine phosphatase for IRS-1 in vitro (Shi et al., 2014). Nonetheless, the finding by Marciniak et al. (2017) that tau has insulin signaling regulator functions in the brain is remarkable, as it further expands the knowledge about AD and brain insulin resistance.
However, some of the results of this study need independent confirmation in other tau KO models. Indeed, the tau KO model used here is not a true KO, as it was produced by insertion of EGFP in exon 1 of MAPT, and a fusion protein with the first 31 amino acids of tau followed by EGFP is expressed (Tucker et al., 2001). Whether some of the results might stem from the production of this fragment in the absence of functional murine tau will need to be clarified. This also raises the question as to which region of tau is mediating the effects observed. The repeat region is involved in microtubules binding and stability, whereas the projection domain mediates some of tau signaling functions (Guo et al., 2017). The region or regions where PTEN binds and the region or regions important for the regulation of brain insulin sensitivity might be the same or might be different.
Similarly, and as mentioned by Marciniak et al. (2017), it would be important to address the sensitivity of central and peripheral insulin in a conditional KO mice model because tau is present in the pancreas and could even modulate the secretion and the transcription of insulin (Neuville et al., 1995; Maj et al., 2016). Peripheral injections of insulin in tau KO mice could provide important elements for the understanding of this new function of tau outside of the brain. Finally, the confirmation of these results in a mouse model that exhibits tau pathology will be necessary to support the hypothesis of tau loss of function involvement in brain insulin signaling impairment in AD, although it is not clear whether the mouse models of tauopathies available have prominent tau loss of function.
To conclude, the study by Marciniak et al. (2017) not only identifies a new function of tau protein as a modulator of brain insulin signaling, but also highlights potential mechanistic explanation whereby alteration of insulin signaling would occur in AD via tau loss of function.





